Home
Home Health
Health Diet & Nutrition
Diet & Nutrition Living Well
Living Well More
MoreAt a rate of half a cup a minute, the kidneys filter unwanted waste from the blood to the bladder with the help of millions of microscopic nephrons. Each nephron has a glomerulus and a tubule; the former collects and filters the blood, while the latter separates the waste bound for the bladder from the nutrients it needs to reabsorb. In people with Fanconi syndrome or FS, a defect in the proximal tubules causes poor reabsorption of nutrients and results in lifelong health issues.
Types of Fanconi Syndrome
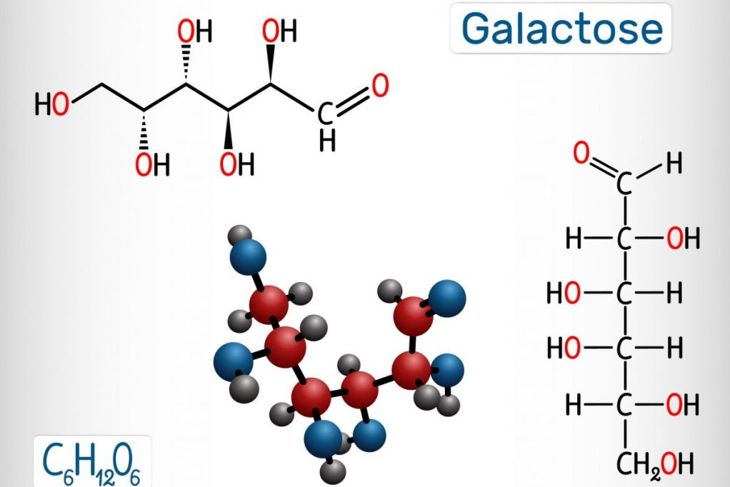
The type of Fanconi syndrome with which a person is diagnosed depends on which nutrients their body cannot reabsorb. For example, galactosemia, the inability of the body to process the sugar galactose, is one manifestation of FS. It can be treated by not giving an affected child milk — lactose breaks down to galactose. Children with this condition will also need to avoid other lactose- and galactose-containing foods. Fructose intolerance and glycogen storage disease may lead to metabolic illnesses, as well.
Genetics
The genetic form of Fanconi syndrome is either X-linked, autosomal recessive, or autosomal dominant. Sometimes, a mutation in the EHHADH gene causes the condition by rendering mitochondria unable to produce ATP, adenosine triphosphate, a chemical that provides energy for all living cells. A person may also have a mutation of the HNF4A or SLC34A1 genes, which are responsible for multiple conditions, including phosphaturia and polyuria.

Cystinosis
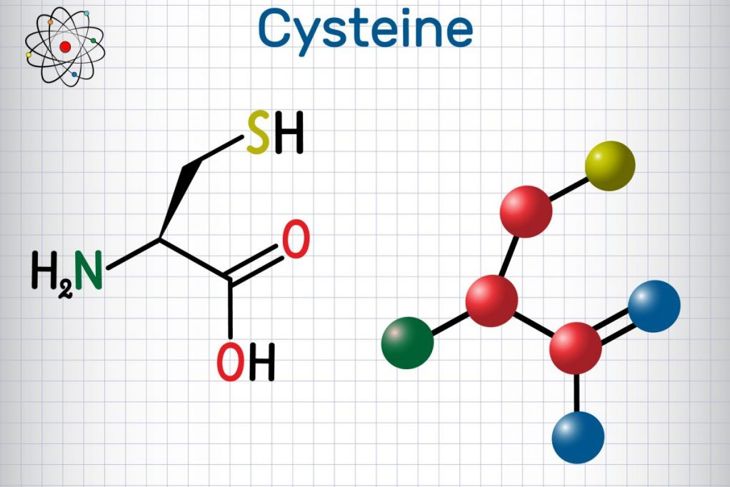
Cystinosis is one of the most common causes of Fanconi syndrome in children and affects about one in every 200,000 births. Individuals with this condition develop a buildup of cystine crystals that damage the kidneys and eyes. Children with this condition may develop kidney failure before their tenth birthday unless the condition is recognized and treated.] Cystine, an amino acid, produces collagen and aids in the synthesis of glutathione, which helps maintain the immune system. There are three types of cystinosis, and each is caused by the mutation of the CTNS gene. CTNS is responsible for directing the production of cystinosin, the transport protein that is key to the digestion and recycling process in the cells. Cystinosis is a genetic disorder inherited in an autosomal recessive manner. So, a person must inherit a copy of the defective gene from each parent to manifest the disease.
Acquired Fanconi Syndrome
A variety of conditions can cause acquired FS, including autoimmune disorders such as Sjogren’s syndrome, which attacks the [glands] joints and kidneys. Heavy metal toxicity, such as lead poisoning, which is more prevalent in children than adults, causes Fanconi syndrome due to the immune system factors that impair tubule function. Drug-related causes include the ingestion of certain antibiotics, including expired tetracycline, that can degrade and damage the proximal tubules. Additionally, drugs that treat dermatological conditions and ulcers may cause FS in some. Other medications linked with this condition include antivirals, chemotherapy agents, and medications used to treat seizures.
Not Fanconi Anemia
Despite having similar names, Fanconi anemia and Fanconi syndrome are entirely different conditions. Guido Fanconi discovered Fanconi anemia in 1927. Also called inherited infantile aplastic anemia, the disorder causes enlarged red blood cells, but low platelet and white blood cell, counts. While children with Fanconi anemia may have malformed kidneys, among other symptoms, it is mostly a disease of the bone marrow, not the kidneys. Estimates indicate that one in 130,000 children is born with the rare disease.
Rickets and Osteomalacia
Both rickets and osteomalacia are related to Fanconi syndrome. A deficiency in calcium, vitamin D, and phosphates causes rickets, leading to bow leggedness and poor bone development. Osteomalacia is softening of the bones caused by the same deficiencies. Examinations of children and adults with Fanconi syndrome showed evidence of rickets and osteomalacia, respectively. Tubular malabsorption of phosphorus and amino acids was the primary cause.

Lowe Syndrome
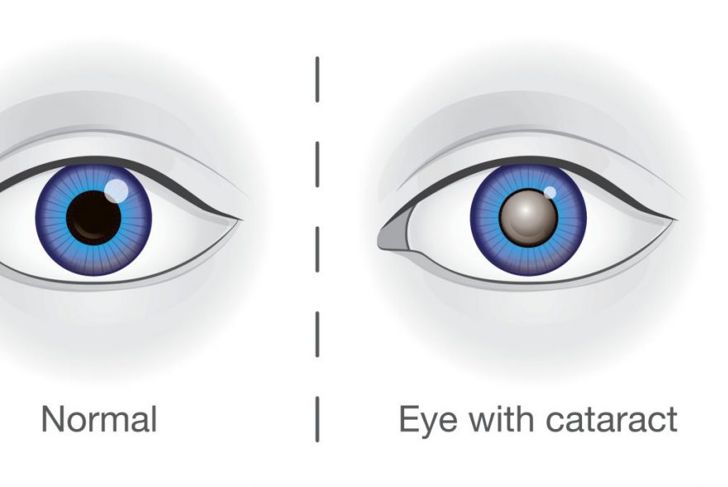
Lowe syndrome is a disorder that affects the brain, kidneys, and eyes. It is X-linked, which means that males are exclusively affected because they lack an additional X chromosome that can compensate. Boys born with Lowe syndrome have weak muscle tone, delayed development, seizures, and eye problems, such as bilateral cataracts or glaucoma. Due to the inherent renal abnormalities, half of all cases develop Fanconi syndrome.
Symptoms and Diagnosis
Fanconi syndrome symptoms depend on the underlying condition, but muscle and bone weakness are inevitable. Excessive thirst, lethargy, delayed growth, and passing so much urine that it leads to dehydration may also indicate the diagnosis. Based on these symptoms and a physical examination, doctors order urinalysis that tests for abnormal levels of amino acids, phosphates, [icarbonate, glucose, and potassium. Fasting blood specimens with low phosphate also prompt physicians to test for FS.

Treatment Options
There is no cure for Fanconi syndrome, so the first line of treatment is to replace what is lost. For children, that means having ready access to water and electrolytes to avoid dehydration and treating the side effect promptly when it occurs. In adults, mitigating mineral loss and dehydration are also priorities. With osteomalacia and muscle weakness, doctors may recommend higher daily doses of calcium and vitamin D that can help the body heal fractures and gain muscle strength. People with more serious cases may require steroid protocols, as well as supplementation with bicarbonate to lower the acidity of the blood.
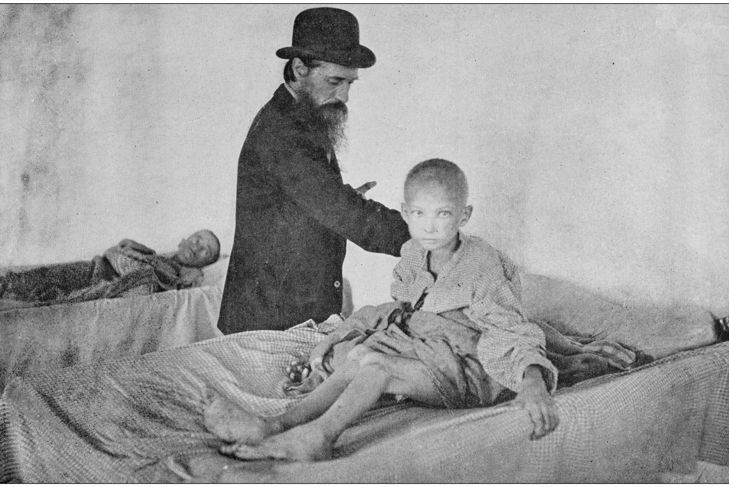
Discovery
While this disease is named after the Swiss pediatrician Guido Fanconi, another Swiss physiologist, Emil Abderhalden, discovered the first case of nephritic cystinosis in a 21-month-old child in 1903. There were separate cases of severe rickets, hypophosphatemia, and albuminuria — all related in cause — between 1903 and 1931. Fanconi recognized the similarities between these cases, which led to more insight into this condition.