Home
Home Health
Health Diet & Nutrition
Diet & Nutrition Living Well
Living Well More
MoreLysosomal storage disorders are caused by an inherited inability to break down certain sugars or fats in the body. When a person has one of these conditions, they don’t produce the enzymes they need to break down these substances at a cellular level. This leads to a build-up of these substances and the development of a lysosomal storage disease. Most lysosomal storage disorders can only develop when a child inherits an abnormal gene from both parents.
Types of Lysosomal Storage Disease
There are many lysosomal storage diseases. Currently, experts have discovered more than 40, with new ones being identified all the time. Different lysosomal diseases cause different symptoms and affect people with varying severity. Common varieties include Gaucher disease, Fabry disease, Hunter disease, and Tay-Sachs disease. Each disorder affects a different enzyme responsible for breaking down a particular substance in the body.

Prevalence
Each lysosomal storage disease is quite rare. However, because there are so many different types, when all the incidences are added up this is quite a common health condition. Currently, around one in 8000 people are born with some form of lysosomal storage disease. However, researchers believe this figure may be lower than the actual incidence rate of lysosomal storage diseases because some people are affected mildly enough that they remain undiagnosed.

Risk Factors
Usually, both parents have to be carriers of the abnormal gene of a lysosomal storage disease for their children to be at risk. The exceptions to this are Hunter disease and Fabry disease. Certain ethnic groups have a higher risk of developing particular lysosomal storage diseases than others. For example, Gaucher disease and Tay-Sachs disease are far more prevalent in people of Ashkenazi Jewish descent than the rest of the population.

Common Symptoms
Lysosomal storage disorders are progressive diseases. This means the individual is usually born healthy and their condition deteriorates over time. Symptoms can affect various systems in the body including the bones and joints, nervous system, circulatory system, and internal organs. Looking at which areas are affected can help doctors diagnose the specific lysosomal storage disorder.

Diagnosis
Because lysosomal storage diseases are rare, diagnosis can be challenging. Sometimes, doctors recognize the symptoms of a particular disorder and can make a diagnosis. However, it is more likely an individual will be investigated for other common conditions before the doctor reaches a correct diagnosis. Tests can screen for certain disorders; for example, blood tests may identify that an enzyme is absent, and biopsies or scans can detect signs of disease.
Treatment Options
While lysosomal storage diseases are incurable, treatments can help manage the symptoms and slow the progress of the condition. Whether there is an effective treatment available depends on the specific lysosomal disorder. In some cases, doctors can replace the missing enzyme intravenously. They may also prescribe drugs to reduce the amount of the unmanageable substance building up in the cells. Occasionally, doctors may recommend a stem cell transplant, which can help the body begin making the missing enzyme itself.
Tay-Sachs Disease
People with Tay-Sachs disease cannot make an enzyme called hexosaminidases A, which breaks down fats in the brain. Lack of this enzyme causes damage to nerves, resulting in slow development, weakness, paralysis, loss of vision and hearing, and trouble feeding. Untreated, the condition can be fatal. However, doctors can treat affected children using medicines and other therapies. The disorder usually starts in infancy, though some children do not develop signs of the disease until later childhood or even adulthood.
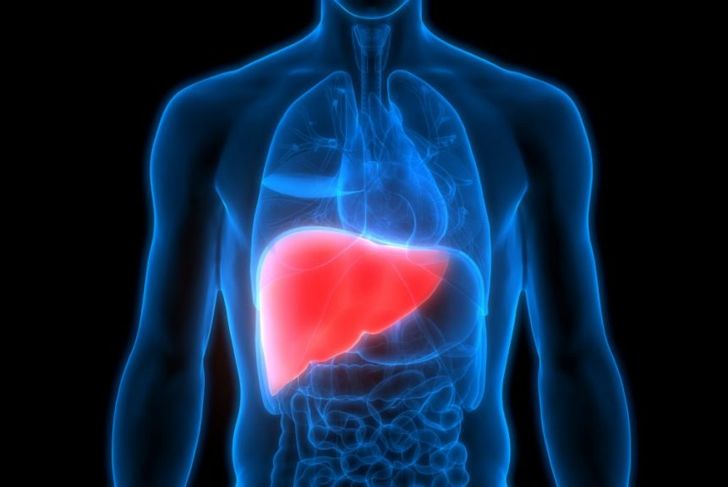
Gaucher Disease
People with Gaucher disease lack an enzyme that ultimately affects the spleen, liver, and bones. Organs become enlarged and function improperly. Insufficient bone development leads to more frequent fractures. Very rarely, Gaucher disease can also affect the brain. In this situation, the condition is fatal. Some people are affected only mildly by Gaucher disease and require no treatment. For those who do, a range of effective treatments is available, including medications and enzyme replacement therapy, and symptom-based treatments. For example, if the individual’s bones are affected, they may require treatment for osteoporosis.
Hunter Syndrome
Hunter syndrome stops the body from effectively breaking down certain sugars. This causes progressive damage including changes to a person’s appearance, intellectual capabilities, and physical ability. The disorder usually becomes apparent in early childhood and affects boys almost exclusively. Unlike most other lysosomal disorders, people with Hunter syndrome inherit the condition from the mother only. Treatment focuses on managing symptoms to improve the child’s quality of life. However, experts are developing new treatments such as enzyme and gene therapy, to slow the progress of Hunter syndrome and lessen the severity of symptoms.

Fabry Disease
Fabry disease affects the body’s ability to break down fatty substances; a lack of this ability narrows the blood vessels. This commonly causes a rash and painful sensations in the hands and feet and can affect various organs in the body. Appropriate treatments can usually control symptoms of Fabry disease. Doctors will prescribe medication or enzyme replacement therapy to prevent further damage to organs.